
Most reviews about ISD (e.g. Draine, 2003a; Whittet, 2003; Tielens, 2005; Draine, 2011; Jones, 2016a,b,c; Galliano et al., 2018) assume that the reader has a good knowledge of solid-state physics 1. It is however not always the case, especially among students. The present chapter is intended to synthesize the basic knowledge necessary to understand contemporary publications in the field. We have opted for a simple presentation of the most important concepts, accompanied with a few original figures. We present the most important formulae and refer the reader to reliable textbooks for their proofs. We have tried to answer everything students always wanted to know about dust, but were afraid to ask.
Atoms can be combined to form molecules or solids. The properties of these compounds depend greatly on the way their constitutive atoms are bonded together, by their electrons. Electrons being fermions (their spin is 1/2), the Pauli exclusion principle implies that each of them must occupy a different state in a system, characterized by its wave function (e.g. Tome II, Chapter XIV of Cohen-Tannoudji et al., 1996). The electronic shells of atoms, the bonds of molecules and the band structure of solids all follow from this principle.
The electrons of an atom each have a distinct wave function, , called orbital. The probability density function of presence of the electron is proportional to . These orbitals are solutions to the Schrödinger equation, in the electrostatic well created by the nucleus, assumed to be infinitely heavy 2. These solutions are relatively simple in the case of the hydrogen atom (cf. e.g. Chap. 3 of Bransden & Joachain, 1983). Its single electron can occupy different shells, characterized by their principal quantum number, . This quantum number determines the energy level of the orbitals () as well as their size (cf. Table 1.1). Each individual shell is divided into subshells, characterized by the azimuthal number, , quantifying the angular momentum of the electron (), which can be 0, for s subshells (cf. Table 1.2). The orbitals in each subshell are combinations of spherical harmonic functions. These functions are anisotropic. The magnetic quantum number, , quantifies their orientation in space, as displayed in Fig. 1.1. Finally, the spin quantum number, , quantifies the direction of the electronic spin (up or down).
| Principal | Bohr | Azimuthal quantum number |
|
|||
| quantum | shells | s | p | d | f | Subshell letter |
| number | 2 | 6 | 10 | 14 | Number of electrons per subshell |
|
| K |
|
|||||
| L |
|
|||||
| M |
|
|||||
| N |
|
|||||
The different energy levels of an atom can be populated by excitation (collision or photon absorption). In the fundamental state of the H atom, the electron occupies the level. The electrons of a polyelectronic atom in its fundamental state occupy the lowest energy orbitals available. Each electron must have a unique set of the four quantum numbers (Table 1.1). The nucleus charge, , is higher, which causes the inner shells to be closer to the nucleus. The electric field seen by the outer shells is thus partially screened by the inner shells. A fundamental difference between polyelectronic atoms and hydrogen is however the mutual repulsion of the electrons (cf. e.g. Chap. 7 of Bransden & Joachain, 1983). The different subshells (s, p, d, f) of a given shell having different geometries, the mutual repulsion depends on . The subshells of a given thus have different energies. The electronic configuration of atoms in their fundamental state results from the ranking in energy of these levels. The possible number of electrons per subshell is given in Table 1.2. We have represented the electronic configuration of atoms in the periodic table (Fig. 1.2).

The outer shell is called the valence shell. It contains the electrons responsible for molecular bonds (cf. Sect. 1.1.2) and shaping the optical properties of solids (cf. Sect. 1.2). We will see that the nature of the chemical bond depends on the tendency of its atoms: (i) to share electrons; (ii) to form cations, by losing one or several electrons; or (iii) to form anions, by gaining one or several electrons.
Ionization potential. Panel (a) of Fig. 1.3 shows the first ionization potential, , of the elements in Fig. 1.2. Atoms with a low tend to form stable cations. Noble gases have the largest of their row, because their last shell is full. More generally, Fig. 1.3 shows that increases from the left to the right of the periodic table (Fig. 1.2), as the valence shell gets fuller. This is because, at a given , moving to the right of the table increases the effective nucleus charge , making the valence shell more tightly bound. It also decreases from the top to the bottom, as the energy level of the outer shell decreases with as .
Metals tend to form stable cations.
Electron affinity. Panel (b) of Fig. 1.3 shows the first electron affinity, , of the elements in Fig. 1.2. Atoms with a high electron affinity tend to form stable anions. follows roughly the same trend as , with some exceptions. Noble gases have their last shell full, they therefore tend to remain neutral and have a negative . Alkaline earth metals also have negative , because of the energy difference between their full s and empty p shells.
Non metals tend to form stable anions.
Electronegativity. The balance between and gives an idea of the tendency of an atom to attract electrons in a bond. Mulliken’s electronegativity, , is defined as (e.g. Huheey et al., 1993):
| (1.1) |
If we except noble gases, we see that will be higher to the right of the periodic table, and will decrease from the top to the bottom.

To explain the shape of molecules, the concept of hybrid orbitals was introduced in the 1930’s by Linus PAULING. The principle is that orbitals with similar energies can be linearly combined together to form new orbitals with different shapes 3. The most relevant example to ISD is the hybridisation of carbon. A 2s electron can be promoted to 2p, resulting in the following configuration.

The carbon is now in an excited state, noted C. The promotion requires 4.2 eV. From this new state, the following combinations of orbitals are possible.



 |  |  |
| (a) sp hybrid | (b) sp hybrid | (c) sp hybrid |
Chemical bonds are the result of the overlap between the outer orbitals of two atoms whose valence shell is not full (cf. e.g. Chap. 8 of Atkins, 1992). Despite their mutual repulsion, sharing electrons leads to a lower energy state, in which a stable bonded molecule is formed. Covalent, ionic and metallic bonds, that we will define below, typically have dissociation energies of a few eV. The atomic spacing in a molecule or a solid is of the order of a few Å.

A covalent bond is formed when a pair of electrons with anti-parallel spin is shared between two atoms of similar electronegativity. The more the orbitals overlap, the stronger the bond is. The electron density is the highest between the two atoms, resulting in a directional bond. For instance, the H2 (Fig. 1.5.a) and CO molecule bonds, as well as the C-C and C-H bonds in hydrocarbons, are all covalent. Covalent bonds are weakly polar. Symmetric molecules such as H2 are non-polar, whereas asymmetric molecules, such as CO are polar, because of the difference in electronegativity of C and O.
Covalent bonds are preferentially formed between non metals.
Covalent bonds are of one of the two following types.
Finally, some transitions in interstellar solids involve antibonding orbitals. When a bond forms, both a bonding and an antibonding molecular orbitals with different energy levels become available. This is demonstrated for the H molecule, with the Schödinger equation, in Chap. 9 of Bransden & Joachain (1983). Bonding orbitals have a lower energy level than the dissociated atoms, thus favoring a stable molecule. On the contrary, the population of an antibonding orbital makes the molecule unstable. For instance, the splitting of molecular orbitals for both and bonds of ethylene (Fig. 1.6) are the following.

We emphasize those are electronic levels of molecules. Molecules also have rotational and vibrational modes that will be discussed in Sect. 1.2.2.1.
 |  |
| (a) bonds | (b) overlap of two p orbitals to form one bond |
An ionic bond is formed between two atoms of significantly different electronegativities. The electron is transferred from the cation to the anion, resulting in a polar bond. The adhesion is due to long-range Coulomb forces () between the two ions. Ionic bonding is non-directional as the electron cloud stays centered around the atoms. The most relevant example to ISD is the O–Mg bond in silicates (Fig. 1.5.b).
Ionic bonds are preferentially formed between a metal and a non metal.
Covalent and ionic bonds are two extreme cases. Most bonds involving at least one non metal are intermediate between both.
Metals can easily be ionized. Bonding several metal atoms therefore results in a lattice of cations bathed in a sea of free valence electrons. The electrons are not attached to a particular atom and can be found anywhere in the solid. This explains the electric and thermal conductivities of metals. Fig. 1.5.c represents solid iron.
Metallic bonds are formed between a large number of metal atoms.
Weaker forms of attraction between molecules exist. Their dissociation energy is typically of the order of eV. They are relevant to ISD studies.
There are two main types of solids: insulators and conductors. Their properties are radically different. Their difference originates in the type of chemical bond making up their crystal lattice.
There is a third, intermediate type of solids, called semiconductors. Semiconductors are insulators at K and conductors at ambient temperatures. Several cosmic dust candidates belong to this category. We will define it more precisely in Sect. 1.1.3.3.
A solid can be idealized as a periodic lattice of atoms bonded to each other. The permitted energy levels of a single valence electron, in the periodic electrostatic potential created by this lattice, are a series of continuous functions, also called bands (e.g. Chap. 8 of Ashcroft & Mermin, 1976, for a derivation from the Schrödinger equation). This can be viewed as a generalization of the molecular level splitting (Fig. 1.7). The spacing between a large number of levels is so small that it appears continuous. At K, the lowest energy bands are filled in priority. Two of these bands are particularly important.
The energy difference between the top of the valence band and the bottom of the conduction band is called the band gap, noted (Fig. 1.7).

The probability distribution of identical fermions, such as electrons in a solid, over the energy states of a system at temperature , is given by the Fermi-Dirac distribution:
| (1.2) |
where is the Boltzmann constant (cf. Table B.2), denotes the different energy levels and is the Fermi level 4. This distribution is displayed in Fig. 1.8.a. The Fermi level is an intrinsic quantity characterizing a solid. It is the energy required to add an electron to the system. It also corresponds to the maximum energy an electron can have at K. The latter interpretation of can be seen in Fig. 1.8.a. The blue curve shows Eq. () at K: (i) it gives equal probability to electrons to occupy energy levels ; (ii) it gives zero probability to energy levels . The actual number density of electrons, , is:
| (1.3) |
where is the density of states per infinitesimal energy bin. This density of states corresponds to the band structure. It is 0 between the bands. We emphasize that can fall between two bands. It does not necessarily correspond to an actual allowed level. This is demonstrated in Fig. 1.8.

We briefly review here the constitution of the most likely ISD grain candidates. Some general properties are given in Table 1.3. Their optical properties are extensively discussed in Sect. 1.2.2.
The different types of silicates are built around silica tetrahedra (SiO), paired with various cations to produce a neutral compound (cf. e.g. Henning, 2010, for a review). The silica tetrahedra have a central Si cation tied to four O anions with covalent/ionic bonds. In the ISM, the most widely available divalent cations that can be paired with silica tetrahedra are Mg and Fe (cf. Sect. 2.2.3). Silicates have two strong features at 9.7 (Si–O stretching) and 18 (O–Si–O bending). They are ubiquitous: (i) they are the main constituent of Earth’s crust; (ii) they are also found in Solar system and CircumStellar Dust (CSD); (iii) they account for probably 2/3 of interstellar grain mass (e.g. Draine, 2003a); (iv) their features are observed in distant galaxies, in absorption (e.g. Marcillac et al., 2006) and in emission (e.g. Hony et al., 2011). Interstellar silicates are widely amorphous (e.g. Kemper et al., 2004). Crystalline silicates have additional distinctive narrow features, due to SiO as well as (Fe,Mg)–O vibrations, in the 9.0–12.5 and 14-22 ranges, with a few bands above 33 . The following two types of silicates are the most relevant to ISD (cf. Table 1.3).
Olivine. Olivine have the general formula (Mg,Fe)SiO, with different proportions of Mg and Fe. Its crystalline structure is represented on Fig. 1.9.b. The two following compounds are the extreme cases of the Fe-to-Mg ratio.
Olivine have an olive green color (Fig. 1.10.a).
Pyroxene. Pyroxene have the general formula (Mg,Fe)SiO, with different proportions of Mg and Fe. They are constituted of silica tetrahedron chains, sharing one O atom (Fig. 1.9.c), which explains their stoichiometry. The two following compounds are the extreme cases of the Fe-to-Mg ratio.
Pyroxene can be darker than olivine (Fig. 1.10.b).
In general, silicates are translucent minerals. They are gemstones, used in jewelry.

 |  |  |  |  |
(a) Forsterite
(b) Enstatite
(c) Soot
a-C(:H)
(d) Graphite
(e) PAHs
This is a broad class of solids, noted a-C(:H) (a notation introduced by Jones, 2012b). Carbon atoms can be paired in the following ways (Fig. 1.9.d).
The hydrogenation of a-C(:H) influences directly their band gap (Jones, 2012b). Generally, H-poor a-C(:H), which can be noted a-C, are sp dominated (aromatic/olefinic), and have a low band gap ( eV). On the contrary, H-rich a-C(:H), which can be noted a-C:H, are mostly aliphatic (sp), and have a larger band gap ( eV). The aromatic domains are responsible for bright features at 3.3, 6.2, 7.7, 8.6 and 11.3 , that will be extensively discussed in Sect. 3.2.1.1, whereas the main aliphatic feature is at 3.4 . An important feature at 2175 Å (Sect. 2.2.1) is thought to originate in the transition between the and bands of sp domains (e.g. Draine & Li, 2001).
a-C(:H) tend to be more opaque than silicates (Fig. 1.10.c).
Graphite is a mineral made of the stacking of graphene sheets, bonded by Van Der Waals interactions (Sect. 1.1.2). Graphene sheets are planar compounds exclusively constituted of aromatic cycles (Fig. 1.9.e). Pure graphite is solely made of sp carbon. Its aromaticity explains its shiny silver metallic appearance (Fig. 1.10.d). It has a strong transition around 2175 Å. The exact central wavelength however depends on the size and shape of the particles, and pure graphite seems too wide to account for the interstellar feature (e.g. Draine & Malhotra, 1993; Voshchinnikov, 2004; Papoular & Papoular, 2009). Graphite also has a broad band at 30 , seen parallel to the sheets, which corresponds to the oscillation frequency of the delocalized electrons (e.g. Venghaus, 1977; Draine & Li, 2007).
PAHs are a class of molecules made of aromatic cycles, with peripheral H atoms (Fig. 1.9.f). They have the aromatic features of a-C, as well as the transition around 2175 Å (e.g. Joblin et al., 1992). Similarly to graphite, the exact central wavelength depends on the particle size and shape (e.g. Duley & Seahra, 1998). They can be colorful (cf. Fig. 1.10.e). They are highly flammable and carcinogenic.
| Name | Stoichiometry | Density | Melting | Main spectroscopic features |
| SILICATES
| ||||
| Forsterite | MgSiO | 3.3 g/cm | 2200 K |
9.7, 18 |
| Fayalite | FeSiO | 4.4 g/cm | 1500 K |
9.7, 18 |
| Enstatite | MgSiO | 3.2 g/cm | 2100 K |
9.7, 18 |
| Ferrosilite | FeSiO | 4.0 g/cm | 1200 K |
9.7, 18 |
| CARBONACEOUS
| ||||
| a-C(:H) | CH | 1.8–2.1 g/cm | N/A |
2175 Å, 3.3, 3.4, 6.2, 7.7, 8.6, 11.3 |
| Graphite | C | 2.3 g/cm | 3600 K |
2175 Å, 30 |
| PAH | CH | 2.2 g/cm | N/A |
2175 Å, 3.3, 6.2, 7.7, 8.6, 11.3 |
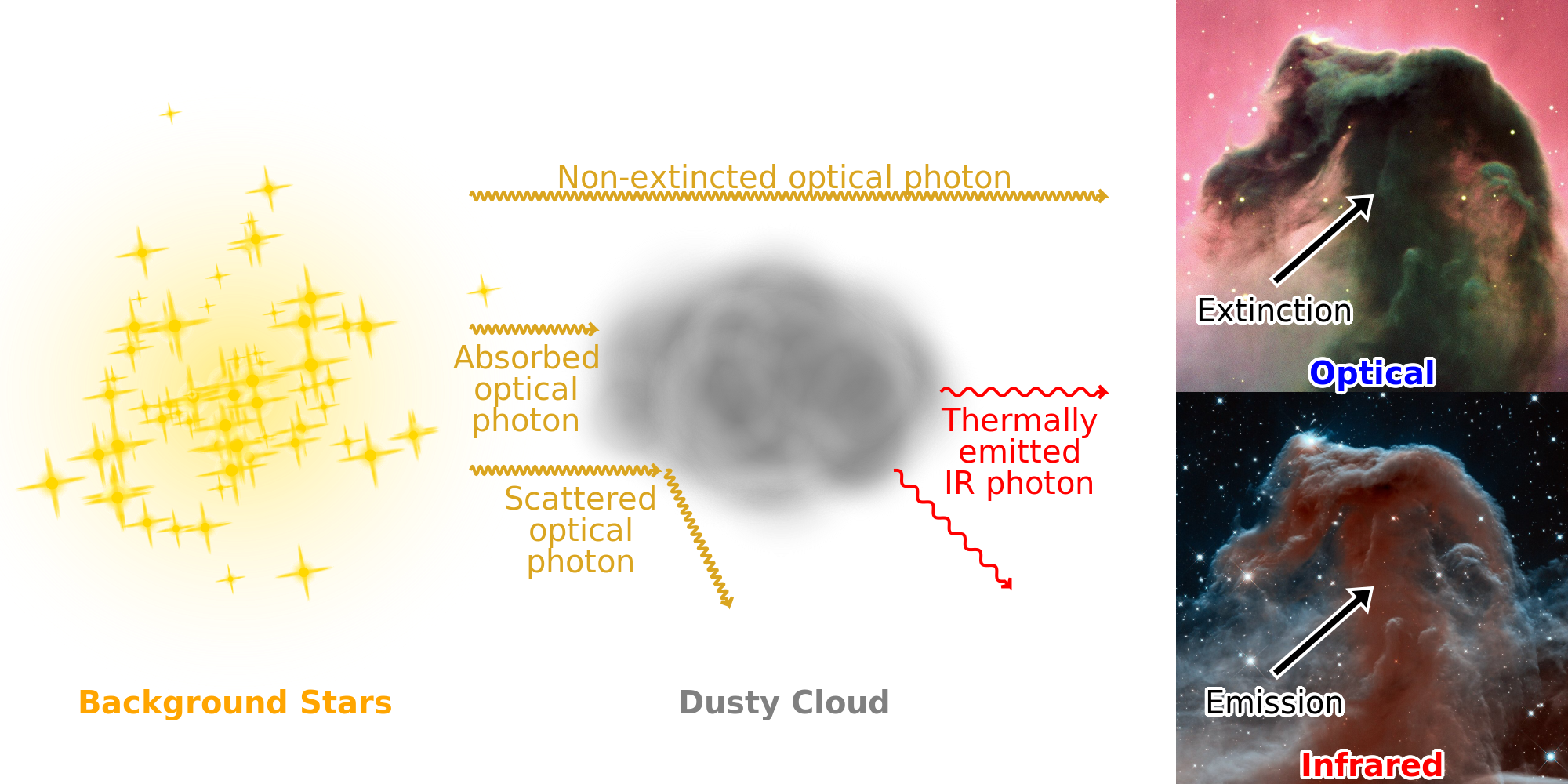
The interaction of an electromagnetic wave with dust grains results in the three following phenomena (cf. Fig. 1.11).
The sum of absorption and scattering is called extinction. These three phenomena can be modeled, assuming valence electrons are harmonic oscillators. This way, the response to an electromagnetic wave of bonds in a dielectric or free electrons in a metal can be quantified. This is illustrated in Fig. 1.12. Throughout this manuscript, we use , and to respectively denote the wavelength, frequency and angular frequency of an electromagnetic wave, being the speed of light (cf. Table B.2).

The harmonic oscillator model is particularly useful to describe the way bonds react to electromagnetic waves.
The position along the -axis of a unidimensional harmonic oscillator of mass, , as a function of time, , follows the equation:
| (1.4) |
where is the external force applied to the oscillator, is the strength of the restoring force, and is a dissipation constant. This equation is simply the Newton law (), where the force, , has three components: (i) the external force, , displacing the oscillator out of its equilibrium position (); (ii) the restoring force, , which is proportional to the distance, meaning it is stronger when the oscillator is farther away from its equilibrium position; (iii) the friction force, , proportional to the velocity, having the effect of slowing down the oscillator.
In the case of the motion of an electron, excited by the Lorentz force of a complex, harmonic plane electromagnetic wave with angular frequency , , Eq. () can be rewritten:
| (1.5) |
where is the electron mass. We have also introduced the natural frequency, , and the damping constant, :
| (1.6) |
In this case, the restoring force is created by the atom’s electrostatic potential well, and the friction can be interpreted as collisions of the electron with the lattice. The solution to Eq. () has the form , with complex amplitude (e.g. Levi, 2016):
| (1.7) |
It is important to consider both and as complex quantities, since there is a phase shift induced by the dissipation term. The module of , giving the physical value of the amplitude, is:
| (1.8) |
This is the classic harmonic oscillator solution. It is represented in Fig. 1.13. The amplitude is maximum at the resonant frequency, . If there is no dissipation (), the amplitude becomes infinite, and the electron escapes.
 |  |
Free electrons oscillate around heavy cations at the plasma frequency, . Its formula is (cf. e.g. Chap. 1 of Krügel, 2003, for a simple proof):
| (1.9) |
where is the number density of free electrons. It applies to metals, as well as actual gaseous plasmas. These media absorb and scatter electromagnetic waves with frequencies lower than . For instance, in the case of the Earth’s ionosphere, m, thus MHz. This explains why amateur radio operators communicate over long distances at frequencies lower than this value, to benefit from the reflection of their transmission on the ionosphere (e.g. Perry et al., 2018). In the case of a metal, with density m, PHz, corresponding to a wavelength , in the UltraViolet (UV; cf. Table A.4). This explains the shiny appearance of metals, as they are able to reflect the visible light, which has a lower frequency than UV photons. It happens that the expression of also appears in the optical properties of dielectrics, and we will use it extensively.
In a dielectric, an electromagnetic wave polarizes the bonds. If we consider each bond as a dipole with moment , the polarization density is defined as:
| (1.10) |
where is the number density of valence electrons. The induced polarization density is directly related to the electric field:
| (1.11) |
where is the electric susceptibility. The electric displacement field, , which accounts for the charge displacement induced by an electric field is defined as:
| (1.12) |
where is the electric permittivity of the medium. The relative electric permittivity, is defined such that: . It is a macroscopic quantity, as no medium is truly continuous. At atomic scales, Eq. () can be broken into two terms:
| (1.13) |
The second equality derives from Eq. (). It also implies that . The induced polarization is, using Eq. () and Eq. ():
| (1.14) |
Finally, a bit of algebra and introducing the plasma frequency (Eq. ), gives:
| (1.15) |
This dispersion relation is called the dielectric function. It is displayed in Fig. 1.14.a. It can be related to the refractive index of the material, . Indeed, plane electromagnetic waves propagating in a dielectric have a phase velocity , where is the magnetic susceptibility (cf. e.g. Chap. 7 of Jackson, 1999, for a derivation from Maxwell’s equations). This phase velocity can also be expressed as a function of the speed of light, : , where is the refractive index. Since we can decompose , and because , we have:
| (1.16) |
The refractive index is sometimes referred to as the optical constants, or simply the “ and ”. In a nonmagnetic medium, , thus:
| (1.17) |
The two complex quantities and contain the same information. Eq. () corresponds to one single type of harmonic oscillator, that is to one mode of one type of bond. An actual dielectric is usually the linear combination of several resonances, such as Eq. (), with different sets of , , and .

The flux carried by a plane electromagnetic wave is given by the time average of the Poynting vector:
| (1.18) |
The power radiated by a dipole, harmonically oscillating, is (cf. e.g. Chap. 9 of Jackson, 1999):
| (1.19) |
This power is the radiation in response to the excitation by the incident wave. This is the scattering contribution. The scattering cross-section of this harmonic oscillator is thus simply: Replacing by Eq. (), we obtain:
| (1.20) |
where we have introduced the Thomson cross-section:
| (1.21) |
Now, the absorbed power comes from the dissipation into the solid. The dissipation force in Eq. () is . The dissipated power is thus the work of this force:
| (1.22) |
The absorption cross-section of the harmonic oscillator is therefore: . Using Eq. () and Eq. (), we obtain:
| (1.23) |
It is interesting to look at the limiting behavior of both and (see also Chap. 1 of Krügel, 2003).
| (1.24) |
| (1.25) |
It shows that around the resonant frequency, both and have Lorentz profiles centered at , with Full Width at Half Maximum (FWHM), .
| (1.26) |
Those approximations are particularly useful.
For dielectrics, and at long wavelength.
Eq. () is valid for a dielectric, as it assumes the medium is only constituted of dipoles. This is not the case in a conductor where there are also free charges. An electromagnetic wave induces a current, , related to the electric field, , by the conductivity, :
| (1.27) |
where the second equality relates the current to its microscopic origin, the velocity of free electrons, . Maxwell’s equations for plane waves give (e.g. Chap. 7 of Jackson, 1999):
| (1.28) |
where is the leftover dielectric term. We can apply the harmonic oscillator model again to these free electrons. The difference is that there is no restoring force, . From Eq. (), the velocity of free electrons becomes:
| (1.29) |
Injecting this quantity in Eq. (), we obtain:
| (1.30) |
Focussing on the free electron term in Eq. (), i.e. assuming , we get:
| (1.31) |
It is also known as the Drude model. This equation is displayed in Fig. 1.14.b. Interestingly enough, the cross-sections of Eq. () and Eq. () apply also to conductors, with . We can easily derive their limiting behavior.
Conductors have the same behavior than dielectrics at long wavelength: and .
The residue theorem implies that, if is a complex function of the complex variable , analytical over , and dropping faster than , we have the relation:
| (1.32) |
with real and positive. We have used the Cauchy principal value:
| (1.33) |
which is simply an integral avoiding the singularity in . Decomposing , we obtain cross-relations between and . These general mathematical relations are usually applied to the susceptibility, from which we derive the dielectric function (e.g. Chap. 21 of Draine, 2011):
| (1.34) |
These relations are known as the Kramers-Kronig relations.
Implications. We only need to specify or at all frequencies, and can use Eq. () to derive the other one. These relations are used to check laboratory data consistency (e.g. Zubko et al., 1996).
Interpretation. They are a consequence of the causality requirement for a linear system (here, we have ). In our case, they impose that the response of the polarization does not precede the effect of the electric field. Sect. 62 of Landau & Lifshtiz (1960), Chap. 21 of Draine (2011), and Chap. 2 of Krügel (2003) discuss these relations more extensively.
Constraint on the Cross-Section. They give some constraints on the long wavelength behavior of the dielectric function. Let’s assume that for , for an arbitrary . The first relation tells us that:
| (1.35) |
The second integral is finite by requirement. For the first integral to be finite, we need to have . At long wavelength, Eq. () tells us that, for a dielectric:
| (1.36) |
We will see in Eq. () that, at long wavelength, . Using Eq. (), we get . We will see in Sect. 3.1.2 that this parameter is sometimes referred to as the emissivity index.
Assuming that at long wavelengths, we need to have .

All but one spectral features of the interstellar grain candidates we have listed in Table 1.3 are in the MIR. This is a general trend (e.g. Table 1 of van der Tak et al., 2018). It can be understood by making an analogy with the different types of molecular transitions (cf. e.g. Chap. 2 of Tielens, 2005, for a review). Those are illustrated in Fig. 1.15.
The dielectric functions of Eq. () and Eq. () correspond to simple cases where there is only one type of oscillator. Realistic materials have more complex structures, with several modes per bond. Deriving dielectric functions of potential interstellar grain analogs is the subject of a rich literature. There are three types of approaches to determine the dispersion relation of a medium.
These approaches are not exclusive and are usually combined as observations and laboratory data are always partial. The work by Draine & Lee (1984) was the first study to use these principles to derive the UV-to-mm dielectric functions of graphite and astronomical silicates. We now have a better knowledge of the dispersion relations of several important materials: (i) silicates (e.g. Laor & Draine, 1993; Weingartner & Draine, 2001a; Draine, 2003b,c); (ii) amorphous carbon (e.g. Rouleau & Martin, 1991; Zubko et al., 1996; Jones, 2012a,b,c); (iii) graphite (e.g. Laor & Draine, 1993; Draine, 2003b,c, 2016); (iv) PAHs (e.g. Li & Draine, 2001; Draine & Li, 2007); and (v) composite grains (e.g. Köhler et al., 2014, 2015). Fig. 1.16 shows a few examples.

The dielectric functions are intensive quantities characterizing the bulk optical properties of solids, but independent of their size and shape. To compute usable absorption and scattering cross-sections, there is one last step to do. Let’s assume our grains are spheres of radius .
| (1.37) |
This expression is simply the geometric cross-section, , times an extinction efficiency, , which is a dimensionless quantity. We can express the scattering and absorption cross-section the same way:
| (1.38) |
Out of these three efficiencies, only two are independent, as we have .
| (1.39) |
| (1.40) |
where is the differential scattering cross-section (i.e. the cross-section for scattering in a given direction), and is the solid angle element. It is normalized over all directions, such that:
| (1.41) |
For isotropic scattering, we have . The first moment of this distribution is called the asymmetry parameter, defined as:
| (1.42) |
This parameter is a direct product of Mie theory. Forward and backward scattering correspond to and , respectively, whereas isotropic scatterers have . There are approximate analytical phase functions. The most famous is from Henyey & Greenstein (1941):
| (1.43) |
Other distributions have been proposed (e.g. Draine, 2003b).
The treatment to compute , and depends on the value of the size parameter:
| (1.44) |
As long as we do not zoom in scales where the hypothesis of a continuous medium breaks down, that is scales of a few Å (i.e. grains made of a few atoms, or hard X-ray photons), the estimation of the efficiencies of a grain only depends on and . The Mie theory (cf. e.g. Chap. 4 of Bohren & Huffman, 1983) 5 is the central tool to compute grain cross-sections. It is a numerical method, exactly solving Maxwell’s equations for the scattering of a plane electromagnetic wave by a homogeneous sphere of known refractive index, . Several regimes can be identified, depending on the value of the size parameter. They are illustrated in Fig. 1.18. In Fig. 1.19, we show the actual cross-sections of silicate and graphite grains of different sizes.
Geometrical optics. It is the regime for which (cf. e.g. Chap. 7 of Bohren & Huffman, 1983). For interstellar grains, which have submicronic sizes, it corresponds to UV wavelengths and shorter. This regime is more relevant to circumstellar dust, where grains can be significantly larger. In geometrical optics, the undulatory nature of light is put aside. Instead, light is modeled as rays, using the formalism of Fresnel. Mie theory is valid in this regime, but numerical problems start arising.

The Mie regime. It corresponds to grain sizes comparable to the wavelength of the incident light (). Mie theory is valid outside this regime, but this is the regime where none of the other approximations are valid.


The Rayleigh regime. It corresponds to grain sizes significantly smaller than the wavelength of the incident light, (cf. e.g. Chap. 5 of Bohren & Huffman, 1983). In the case of interstellar grains, it applies essentially to the NIR regime and longward. The refraction index needs to be small, too: . The solutions are analytic (e.g. Li, 2008):
| (1.45) |
The absorption cross-section of most interstellar grains, in the NIR-to-mm window, is proportional to their volume. The dust mass can thus be probed by absorption or emission measures.
Mie theory is restricted to homogeneous spheres. However, there are several observational indications that this hypothesis is not fully accurate (cf. Sect. 4.2.1.1).
There are several methods to estimate cross-sections of grains beyond the hypothesis of homogeneous spheres.
Effective medium theory (EMT). EMT is a class of methods to replace the individual dielectric functions of a composite material by an average, . Cross-sections can then be estimated using Mie theory or any other approximation. EMT assumes that the different domains are smaller than the wavelength and well-mixed in the grain. There are different mixing rules (cf. e.g. Chap. 8 of Bohren & Huffman, 1983). It seems that their accuracy depends on the type of sample they are applied to, as independent studies find better agreements with one or the other (e.g. Abeles & Gittleman, 1976; Perrin & Lamy, 1990).
| (1.46) |
with:
| (1.47) |
| (1.48) |
Ellipsoids in the Rayleigh regime. They have general analytical solutions (e.g. Chap. 5 of Bohren & Huffman, 1983). When the three axes of the ellipsoid are aligned on the coordinates and if the the electric field is along , we have:
| (1.49) |
where is the shape factor. Noting the two lengths, oblate spheroids have dimensions along the three axes , whereas prolate spheroids have dimensions (cf. Fig. 1.20). With these notations, the shape factor is (e.g. Chap. 22 of Draine, 2011):
| (1.50) |
with . In case of randomly oriented ellipsoids, we simply need to take the arithmetic mean of the cross-sections along the three axes.
 | |  |
| (a) Oblate | (b) Prolate | (c) Mnemotechnics |
Discrete Dipole Approximation (DDA). DDA (Purcell & Pennypacker, 1973) 6 allows the user to model complex composite grains as arbitrary arrays of independent domains. These domains are approximated by a series of discrete dipoles, which must be much smaller than the incoming wavelength. This method is computer intensive, but very flexible (cf. e.g. the results of Köhler et al., 2015; Ysard et al., 2018). We show some of the results of Köhler et al. (2015) in Fig. 1.21. This figure exhibits an important result we will discuss later:
the addition of mantles and the aggregation of grains tend to increase the absorptivity per unit mass in the FIR window.
 |  |
The real part of the electric field of a monochromatic, plane electromagnetic wave, propagating along the -axis, can be written at time and at :
| (1.51) |
where (cf. e.g. Chap. 2 of Bohren & Huffman, 1983). This is the parametric equation of an ellipse in the plane (cf. Fig. 1.22.a). It is the most general case, called elliptical polarization. It is fully characterized by the modules of and , the angle and the rotation direction (-1: clockwise; +1:counterclockwise). Particular cases are: (i) linear polarization, if or ; and (ii) circular polarization, if .
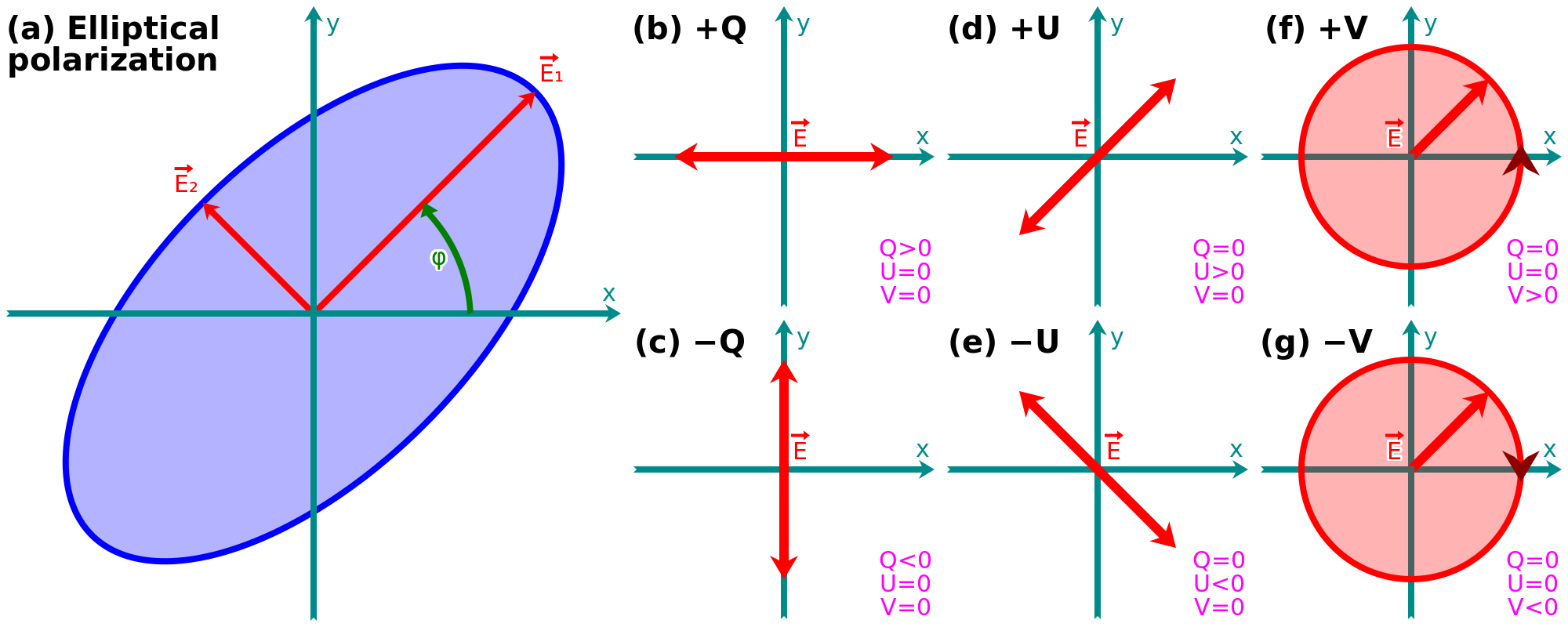
The Stokes parameters. They constitute a four element vector easier to observationally measure than the ellipse parameters. They are noted . is the total intensity of the beam, that can be partially polarized. The three others can be expressed as:
| (1.52) |
The interpretation of these parameters is illustrated in Fig. 1.22. They allow us to decompose the light into its linear and circular polarization components. The intensity of the polarized light is . It is also frequent to quote the linearly polarized intensity:
| (1.53) |
and the linear polarization fraction, .
Grain alignment. Asymmetric grains tend to be aligned with the magnetic field. If we consider the simple case of spheroidal grains (cf. Fig. 1.20): (i) the rotation axis of oblate grains is along their symmetry axis; (ii) the rotation axis of prolate grains is perpendicular to their long axis, their cross-section thus needs to be integrated over their spinning dynamics (e.g. Guillet et al., 2018). The rotation axis of the grains tends to align with the magnetic field, . This is represented in panels (b) and (c) of Fig. 1.23. Several mechanisms have been proposed to explain this alignment (cf. Andersson et al., 2015, for a review). Nowadays, Radiative Alignment Torques (RAT; Dolginov & Mitrofanov, 1976) are favored, because they provide the best account of the observational constraints. This complex scenario is based on the fact that irregular grains have different cross-sections for clockwise and counterclockwise circularly polarized light. Light scattering on such grains therefore provides a torque that increases the angular momentum of the grains. If these grains have paramagnetic inclusions (such as iron), the grain rotation precesses and aligns with . Although the alignment is caused by the radiation field, it is independent of its direction. This mechanism becomes inefficient at high optical depth, which is consistent with observations.
Polarization by scattering. It is represented on Fig. 1.23.a. When an incident beam is scattered by a grain (this grain does not have to be asymmetric), the electric field component in the scattering plane is diminished, inducing a polarization perpendicular to the plane. The larger the scattering angle is, the larger the polarization. The polarization of an incident Stokes vector, , resulting in a scattered beam, , is described as: , where is the Müller matrix (cf. Chap. 3 of Bohren & Huffman, 1983, for different examples of Müller matrices). This polarization process is not related to grain alignment with the magnetic field, but it depends on the distribution of stars and dust clouds (e.g. Wood, 1997).
Dichroic extinction. It is the selective extinction of the electric field oscillating along the major axis of an elongated grains. It is represented in Fig. 1.23.b. Since grains in the diffuse ISM tend to align their rotation axis with the magnetic field, they polarize starlight parallel to . For this reason, the polarization of starlight has historically been used to map the magnetic field of the MW (Mathewson & Ford, 1970).
Polarized emission. The polarization of the emission by elongated grains has been predicted by Stein (1966). Such grains emit IR light preferentially polarized along the direction of their major axis. Since their major axis is perpendicular to the magnetic field, their IR emission is perpendicular to . It is represented in Fig. 1.23.c. The FIR polarized emission has been extensively used to map the magnetic field in the MW, with the Planck satellite (e.g. Planck Collaboration et al., 2016d).
Grain polarization is parallel to the magnetic field in the visible and perpendicular in the IR: and .

In Sect. 1.2.1, presenting the harmonic oscillations of valence electrons, we argued that the energy dissipation was due to collisions with the lattice. The energy absorbed by the grain is thus redistributed throughout the lattice and stored in the harmonic oscillations of its atoms.
The energy levels of a one-dimensional quantum harmonic oscillator, of natural frequency , are (e.g. Chap. 2 of Atkins & Friedman, 2005):
| (1.54) |
where is the Planck constant (cf. Table B.2). At thermal equilibrium, the probability distribution of a large ensemble of such harmonic oscillators, at temperature , is the Boltzmann distribution:
| (1.55) |
where we have introduced the partition function:
| (1.56) |
The second equality comes from injecting Eq. () into the first equality. The mean energy of this ensemble of oscillators is the first moment of the distribution in Eq. ():
| (1.57) |
Since each one of the atoms of the lattice has three degrees of freedom (along the three Cartesian axes ), the number of oscillators is 7. The internal energy of the grain, which is the energy stored in all its oscillators, is thus:
| (1.58) |
Eq. () considers that each atom oscillates independently of its neighbors. In reality, there are collective vibrational modes in a crystal lattice. These modes actually are sound waves, propagating at the sound speed of the material, . Because the size of a grain is finite, the number of these possible modes is quantified. Indeed, if is the size of the grain along one dimension, the wavelength of the modes along this dimension is , with (cf. Fig. 1.24). The shortest possible wavelength corresponds to oscillations of adjacent atoms in opposition of phase: , where is the interatomic distance. These quantified modes can be treated as quasi-particles, called phonons. These phonons have energies , thus:
| (1.59) |
We now need to integrate Eq. () over the different modes:
| (1.60) |
where is the density of modes with frequency , and is a constant coming from the 1/2 term in Eq. (). It can be shown that (cf. Chap. III.E of Diu et al., 1997):
| (1.61) |
where the Debye frequency can be explicited as a function of the density of atoms, :
| (1.62) |
From , we can also define the Debye temperature, . Eq. () thus becomes:
| (1.63) |
and the Debye heat capacity can be derived:
| (1.64) |
It is represented in Fig. 1.25.a.
| (1.65) |
It is the classical expression of heat capacity. It is constant becomes it assumes that energy can be indifferently stored in oscillators, ignoring their limited number.
| (1.66) |
It accounts for the quantification of the modes. It provides a correct agreement with laboratory measurements.


The Debye model is an idealization providing a good approximation. It has however several limitations.
| (1.67) |
where is the Fermi temperature (Eq. ).
Let’s consider a grain at thermal equilibrium with a radiation source, such as the light from a star. The specific intensity received by the grain, , is the electromagnetic power per unit frequency, area (A) and solid angle () 8: (we discuss this quantity in more details in Sect. 3.1.1). The absorption coefficient of this grain, , is the fraction of this specific intensity it absorbs per unit length, : . The emission coefficient of this grain, , is the power it emits per unit frequency, volume (V) and solid angle (it is isotropic): . Kirchhoff (1860)’s law states that the ratio is a universal function depending only on and (e.g. Robitaille, 2009, for a review). Planck (1900) later gave an analytical expression of this empirical function, assuming the energy levels were discrete, providing a quantum formulation of the black body radiation. It became the Planck function, , where:
| (1.68) |
Opacity. The mass absorption coefficient of a grain, , is its absorption cross-section per unit mass: . For a single spherical grain, it is, using Eq. ():
| (1.69) |
This quantity is often referred to as the opacity. In this manuscript, we however extend this term to its scattering component, too. We will therefore call , the opacity, and being called the absorption and scattering opacities, respectively. We have seen in Sect. 1.2.2 that is practically independent of radius for most interstellar grains in the NIR regime and longward, thus:
the NIR-to-mm opacity of interstellar grains having the same homogeneous composition is independent of their radius.
Emissivity. The emissivity of a grain, , is the power it emits per unit frequency and mass (): . The last differential element simply denotes an average over solid angles. We thus see that . Kirchhoff’s law then becomes:
| (1.70) |
Eq. () is the emission spectrum of grains at thermal equilibrium with the radiation field. We show a few examples in Fig. 1.26. The limiting behavior of Eq. () when is given by the Rayleigh-Jeans law:
| (1.71) |

Modified Black Body (MBB). A MBB is an idealized body, at thermal equilibrium with the radiation field, that does not perfectly absorb all frequencies. It is an imperfect black body, sometimes also called grey body. Eq. () is a MBB. In the ISM literature, MBB usually refers to the case where we approximate the opacity as a power-law:
| (1.72) |
where is a reference wavelength, and , the opacity at . This approximation was popularized by Hildebrand (1983). We have seen in Sect. 1.2.1 that for typical grains, and that we must have in order to satisfy the Kramers-Kronig relations (cf. Sect. 1.2.1.6). Eq. () implies that , in the Rayleigh-Jeans regime. We have shown this relation in Fig. 1.26.
| (1.73) |
where is the Stefan-Boltzmann constant (cf. Table B.2). In the case of a MBB, the emitted power per unit mass is:
| (1.74) |
where is the gamma function, and is the Riemann zeta function.
| (1.75) |
where is the Lambert W function.
Eq. () expresses the general emissivity of a grain at thermal equilibrium with the radiation field. To use this formula, we need to determine the equilibrium or steady-state temperature of the grain. This is simply performed by equating the absorbed and emitted powers. It is convenient to define the mean intensity of the ISRF:
| (1.76) |
which is simply the specific intensity from the stars, averaged over solid angle. The power absorbed by the grain is:
| (1.77) |
Similarly, the emitted power is:
| (1.78) |
The equilibrium temperature, , is then simply the numerical solution to . Several quantities can be precomputed to simplify this task.
Planck average. The Planck average of a grain is defined as:
| (1.79) |
This quantity needs to be computed only once for a range of temperatures. Eq. () then simplifies: . Since interstellar grains emit predominantly in the IR, where the approximation of Eq. () is valid, we can derive an analytical expression of Eq. (), knowing that :
| (1.80) |
We show the Planck average of typical grains in Fig. 1.27. We can see that is almost independent of radius for grains smaller than .

ISRF-averaged efficiency. The ISRF-averaged efficiency is the equivalent of the Planck average for the absorbed power:
| (1.81) |
where . This quantity is less general than Eq. () as it needs to be evaluated for each particular shape of the ISRF. Eq. () then simplifies: . The equilibrium temperature is thus the solution to: .
The diffuse ISRF. The diffuse ISRF of the MW has been modeled by Mathis et al. (1983). It is represented in Fig. 1.28. This ISRF is commonly used to describe grain heating (i.e. how much power a grain absorbs) in the MW, and also in nearby galaxies. Most of the heating is provided by the stellar component, as the integrand in Eq. () is . The long wavelengths have a negligible weight. This stellar ISRF can be scaled by a dimensionless factor , to account for variations of the stellar density. This scaling factor is not totally realistic, as regions with high radiation densities () usually are star-forming regions, containing young star associations. The UV bump of the stellar ISRF, corresponding to these young stars in Fig. 1.28, would be dominating the emission, while the NIR bump, corresponding to older stellar populations, would be, at first order constant. This is however not very important for grains at thermal equilibrium, as their spectrum depends only on the total absorbed power, given by .

Equilibrium temperatures. The equilibrium temperature of typical grains is shown in Fig. 1.29, as a function of . Assuming the heating is solely provided by the stellar component in Fig. 1.28, and . In this case, the integrated mean intensity is:
| (1.82) |
The equilibrium temperature is thus:
| (1.83) |
For grains smaller than , we thus have . We show the equilibrium temperature of graphite and silicates, varying in Fig. 1.29. We see that:
Interstellar grains of a given homogeneous composition, at thermal equilibrium with the radiation field, mostly have the same temperature.

Absorption and cooling times. Not all grains are at thermal equilibrium with the ISRF. To determine if this is the case, we need to estimate the photon absorption rate of the grain:
| (1.84) |
where the proportionality has been derived using Eq. (). The grain absorption timescale, , gives the average time between two photon absorptions. We also need to estimate the cooling rate of the grain:
| (1.85) |
where is the enthalpy of the grain at temperature :
| (1.86) |
It is the vibrational energy content of the grain. The proportionality of Eq. () has been derived from Eq. () for , and from the low-temperature behaviour of (Eq. ; corresponding to the three-dimensional Debye model). The cooling time, , is independent of the grain size.
The temperature fluctuations. If , the grain has the time to significantly cool down between two photon absorptions. Its temperature is thus changing with time. This is represented on the simulation in Fig. 1.30.a, as follows.
Inpractice, we do not see the emission of the grain varying with time, as observations encompass a statistical number of grains, with different time histories. What we observe is an ergodic 10 average: small grains appear to have a temperature distribution, represented in Fig. 1.30.b for nm. When the radius of the grain increases, the absorption time decreases as , as shown in the remaining panels of Fig. 1.30. The number of temperature spikes increases, as the grain being larger, it intercepts more photons. The temperature of the spikes also decreases with , as the grain stores the energy of a single photon in a larger number of phonon modes. For large enough grains (Fig. 1.30.g), the temperature fluctuations become negligible. The grain appears to have a single temperature; its temperature distribution tends toward a Dirac distribution (Fig. 1.30.h): it has reached thermal equilibrium. Fig. 1.31 shows the same type of simulations, fixing the radius of the grain, and varying the starlight intensity. In this case, the heating rate increases linearly with (downward in Fig. 1.31).


Out-of-equilibrium emission. At each time, in the simulations of Figs. 1.30 – 1.31, the emission of the grain is: . To average over time, we simply need to integrate over the temperature distribution:
| (1.87) |
The left panels of Fig. 1.32 show the temperature distributions of PAHs, graphite and silicates of different sizes. The corresponding emission spectra, computed using Eq. (), are shown in the right panels. We see that the smallest grains fluctuate to the highest temperatures. Their emission is thus the broadest, and extends to the shortest wavelengths. An important difference between equilibrium and stochastic heating is that the emission spectrum of small grains depends not only on the intensity of the ISRF, but also on its hardness. The latter can be quantified with the mean energy of stellar photons:
| (1.88) |
This parameter determines the average temperature spikes. This is the reason why stochastic heating is sometimes referred to as single photon heating, transient heating or quantum heating. The ISRF intensity roughly scales with the emissivity, but does not affect its spectral shape.
 |  |
Numerical methods. To compute the emission spectrum of an out-of-equilibrium grain, we need to calculate its temperature distribution, (Eq. ). This is done numerically. Several methods can be found in the literature.
Equilibrium criterion. A simple criterion to determine if a grain is at thermal equilibrium with the ISRF consists in comparing its equilibrium enthalpy with the average energy of a stellar photon. Fig. 1.33 compares these two quantities for graphite and silicates, varying and .
A grain is at thermal equilibrium if .
In addition to photon absorption, collisions with gas particles can, in specific conditions, contribute to dust heating. Obviously, this will happen when the temperature of the gas is the hottest, such as in a plasma. The collision rate of gas particles, following a Maxwell-Boltzmann distribution, with a dust grain is:
| (1.89) |
where is the density of the gas, and , the mass of the particles. Assuming that the protons and the electrons are thermalized, the ratio of their collision rates is (cf. Table B.2). The collisions with the protons can thus be neglected.
Electronic heating rate. The Maxwell-Boltzmann distribution of the electrons of energy, , can be written:
| (1.90) |
This distribution is normalized: . It is displayed in Fig. 1.34 and compared to the stellar radiation field. The collisional cross-section is very poorly constrained. At low energies, it should be close to the geometric cross-section, . However, at high energies, electrons can pass thought the grain. Dwek (1986) proposed the following cross-section:
| (1.91) |
where is the threshold energy. According to the fit of experimental data shown in Fig. 1 of Dwek (1987), this threshold energy is (cf. the discussion in Bocchio et al., 2013):
| (1.92) |
Assuming the grains are at rest, the collision rate for a given energy, , is now:
| (1.93) |
where the velocity of an electron with energy is:
| (1.94) |
The collisional power received by the grain is finally the integral of the energy deposit per unit time, , over the whole Maxwell-Boltzmann distribution:
| (1.95) |

Cases where collisions dominate the heating. Coronal plasmas, that can be found in the Hot Ionized Medium (HIM; cf. Table 3.6) of the MW or the halo of elliptical galaxies, have typical temperatures of K. This gas has been heated by successive SuperNova (SN) blast waves. It has a low density ( cm), but can have a large filling factor (it fills of the volume of the MW). At these temperatures, it is responsible for a diffuse X-ray emission. When dust grains are present in such a gas, collisions can dominate the heating, depending on the balance between the photon and electron energy densities (cf. Fig. 1.34).

1.The book by Krügel (2003) is a notable exception. It starts from elementary electrodynamics and atomic physics.
2.The electron-to-proton mass ratio is (cf. Table B.2).
3.The Schrödinger equation is linear. Any combination of solutions is a solution.
4.In the general Fermi-Dirac distribution, the Fermi level, which is proper to solids, is replaced by the chemical potential of the system, . In our case, the Fermi level is the chemical potential of an electron.
5.See also B. Draine’s public code, bhmie.f, implementing the algorithm in Appendix A of Bohren & Huffman (1983).
6.See the public code DDSCAT by Draine & Flatau (1994).
7.This is for . The actual number of degrees of freedom is 3N-6, subtracting the three translatory and three rotational possible motions of the grain as a whole.
8.Throughout this manuscript, we use the subscript to exclusively denote spectral densities, that is quantities per unit frequency, . Such a quantity can also be expressed per unit wavelength: . Quantities depending on the frequency, but not per unit frequency, should not be written with subscript : .
9.In general, we prefer displaying quantities than simply or , as it represents better the energy balance.
10.The ergodicity is a principle stating that the steady-state statistical distribution of the properties of a large number of identical particles is the average of their properties over time.
{kind=link}
{kind=link}
{kind=link}
{kind=link}